Peach (Prunus persica L. Batsch)is a globally important temperate deciduous fruit tree.Because of the long history of peach domestication and improvement as well as a small genome size, peachisconsidered a good model system for studying the interaction between plants and the environment.In 2013, the peach genome wassequenced and released by an international initiative.It was subsequently improved in 2017, whichincreased research interestfor domestication events and gene identification.In the aforementioned studies, all the sequences were aligned to the reference genome, which had been assembled fromthe peach rootstock Lovell. Notably, it is unclear whether this had an impact on the discovery of key genes in cultivated types.
In the present study, an elite landrace, Chinese Cling, was selected for genome sequencing toassemble a chromosome-level genome. The eight pseudo-chromosomes covering the genome with a scaffold N50 of 29.68 Mb, which is 165 gaps lower than that of the previous reference genome.
We compared it with the genome of Lovell to identify variations between these two genomes.A total of 685407 single nucleotide polymorphisms(SNPs), 162655 insertions and deletions(indels), and 16248 structural variantswere identified (Figure 1). A total of 305 and 1 200 gene families were expanded and contractedin the Chinese Clingcompared with other 9 genomes, respectively. The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis indicated the majority of expanded gene families were enriched in selenocompound metabolism and proteasome genes,whereas contracted gene families were enriched in flavone, flavonol, flavonoid, and monoterpenoid biosynthesis genes.
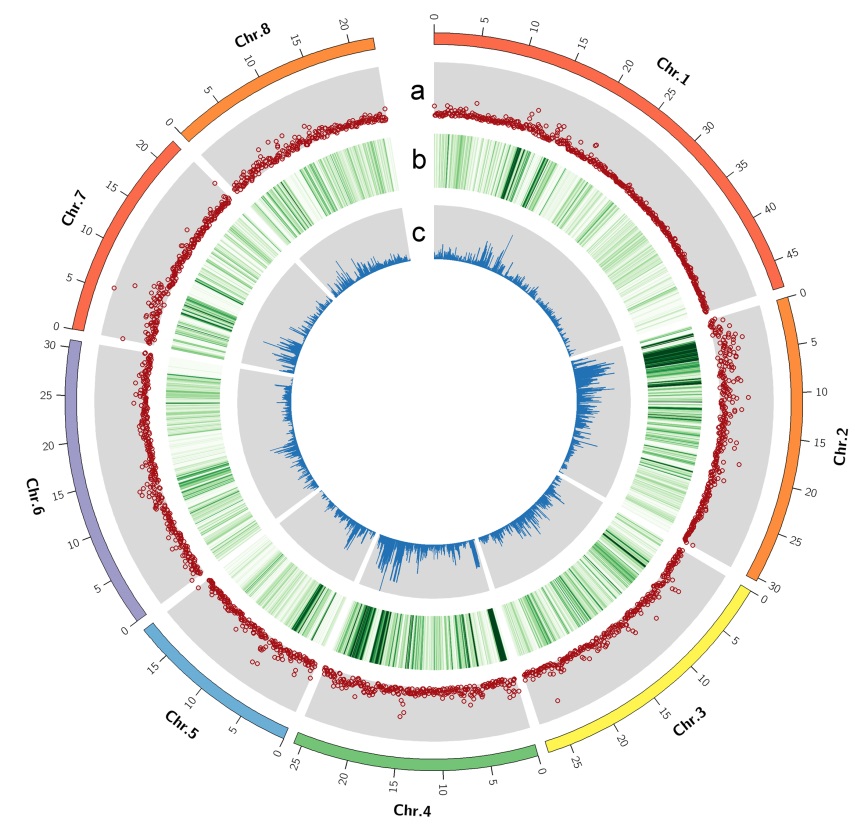
Figure 1 Variations identified by two assembled genomes across the pseudo-chromosomes by Circos.
Moreover, in order to verify the effect of assembly, a GWAS of the traits related to volatile substance content was conducted to discover the key genes for the main aroma components in peach fruit. The GWAS generated 83 and 131 QTLs, associated with 23 of the 28 volatiles of germplasm collections evaluated in 2015 and 2016, respectively. We found that 25 QTLs that were associated with seven volatile compounds overlapped with domestication regions (Figure 2).
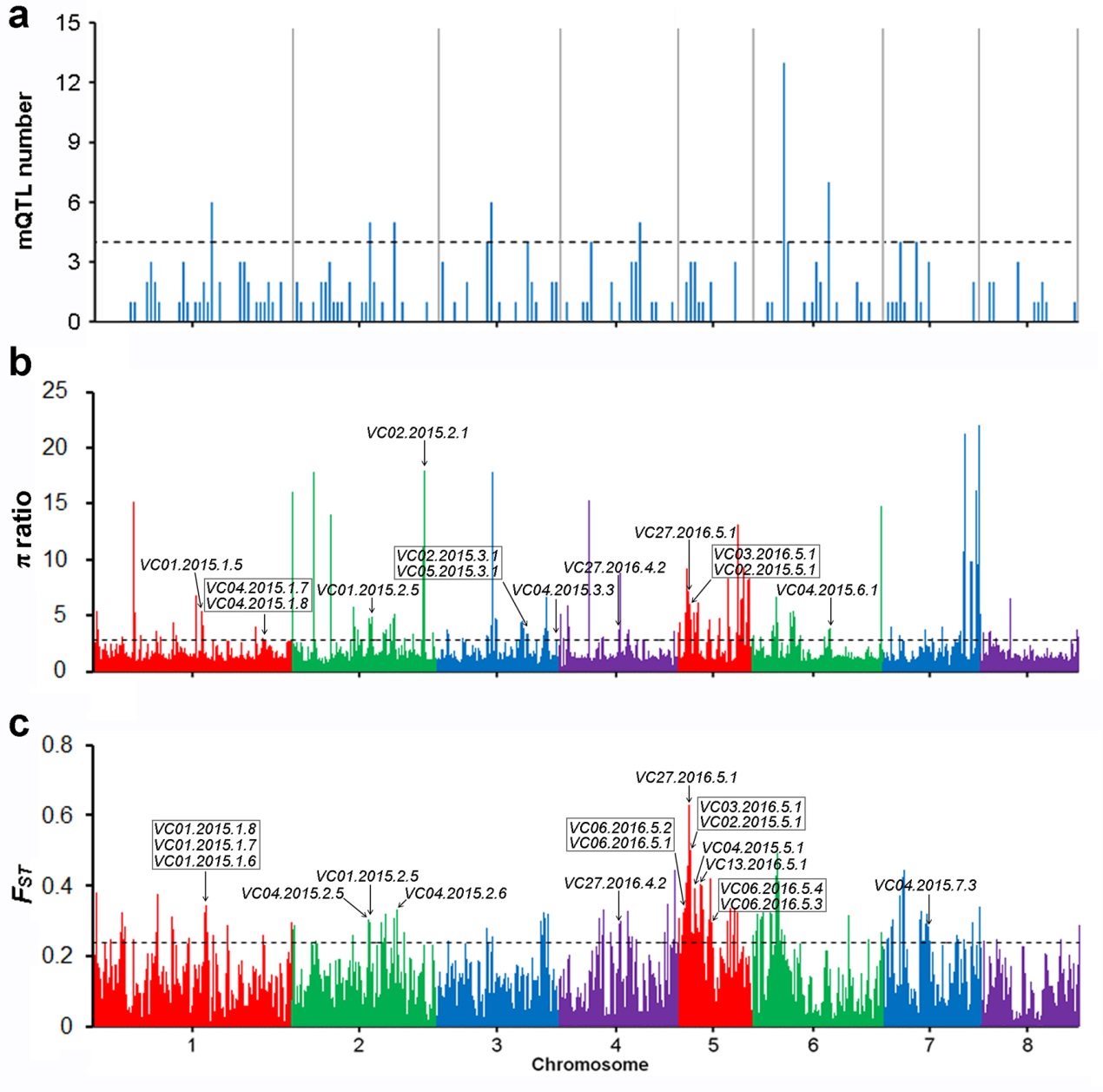
Figure 2 The domestication of volatile compounds in peach fruit.
Among all the volatile substances, the monoterpene linalool is recognized as a major contributor to aroma and flavor in peach fruit. Notably, the association signal of the aromatic compound was repeatedly identified in 2015 and in 2016. Within the flanking regions of association regions of linalool content, Pp4G000350 whichencoding terpene synthasewas considered to be a key candidate gene for linalool content after screening the expressed genes during fruit development and genetype analysis (Figure 3).
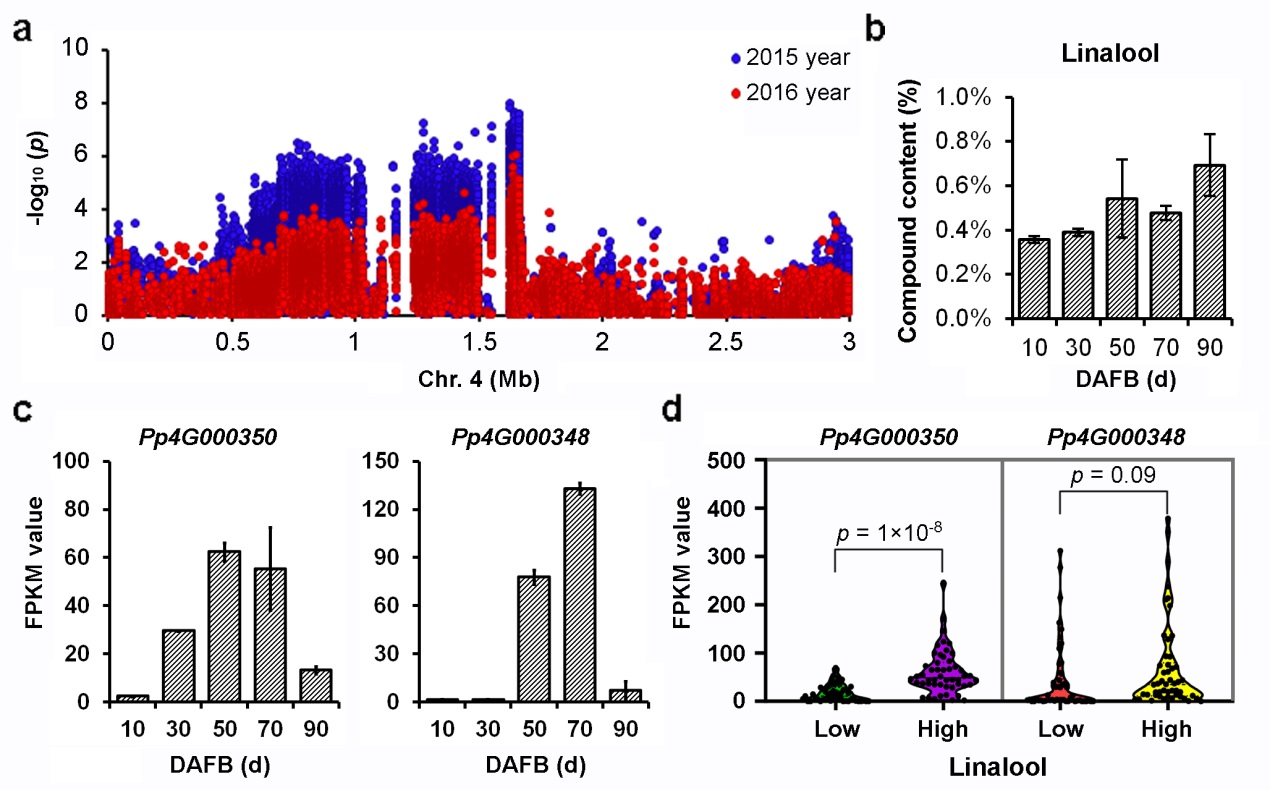
Figure 3 Association signals of linalool content in Chromosome 4 and candidate gene identification.
This study presents a new high-quality peach genome, which should be a useful resource for studying the molecular mechanism of peach domestication and discovering candidate genes related to various traits.This work was supported by the National Key Research andDevelopment Program of China (2019YFD1000200) and the Agricultural Science and Technology Innovation Program of the ChineseAcademy of Agricultural Sciences(CAAS-ASTIP-2016-ZFRI-01).
By Cao Ke
E-mail: wyandck@126.com