Globally, commercialized plum cultivars are mostly diploid Chinese plums ( Prunus salicina Lindl.), also known as Japanese plums, and are one of the most abundant and variable fruit tree species. Recently, the Plum and Apricot Breeding Group and the Peach Germplasm Research Group of Zhengzhou Fruit Research Institute (ZFRI) presented a high-quality chromosome-scale P. salicina genome assembly of cultivar ‘Zhongli No.6’ selected by ZFRI, constructed using an integrated strategy that combines Illumina, Oxford Nanopore, and high-throughput chromosome conformation capture (Hi-C) sequencing, which will provide valuable genetic basis for the improvement of economically important traits of Chines plum. The related results have been published in the Plant journal on September 2, 2021.
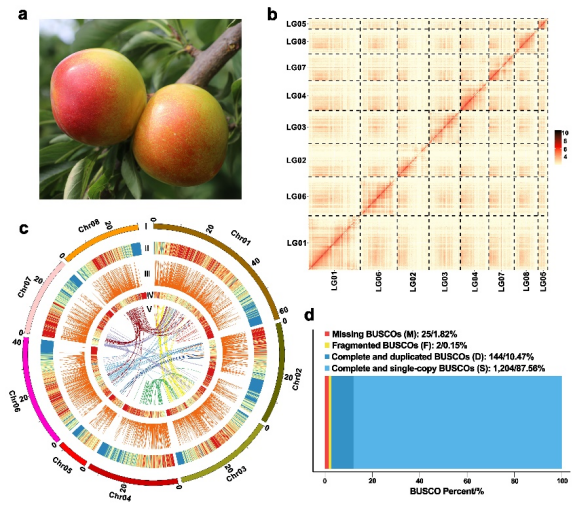
Fig. 1 The genome features of P.salicina.
Southwest China was previously assumed to be a primitive domestication center of Chinese plum, which was evidenced by the discovery of wild plum communities in the middle reaches of Yangtze River Basin, a palynology investigation, and a large-scale phenotypic variation analysis. Using the high-depth (~20 ×) whole-genome re-sequencing data of the 74 accessions, we obtained 3,380,659 high-quality SNPs, which helped classify these accessions into four geographical groups: the South China group, the North China group, the Northeast China group, and the Foreign groups. The Chinese plums in South China possess a broader genetic background and more frequent gene exchange with those from other geographical groups, and their possible center of origin is in South China.
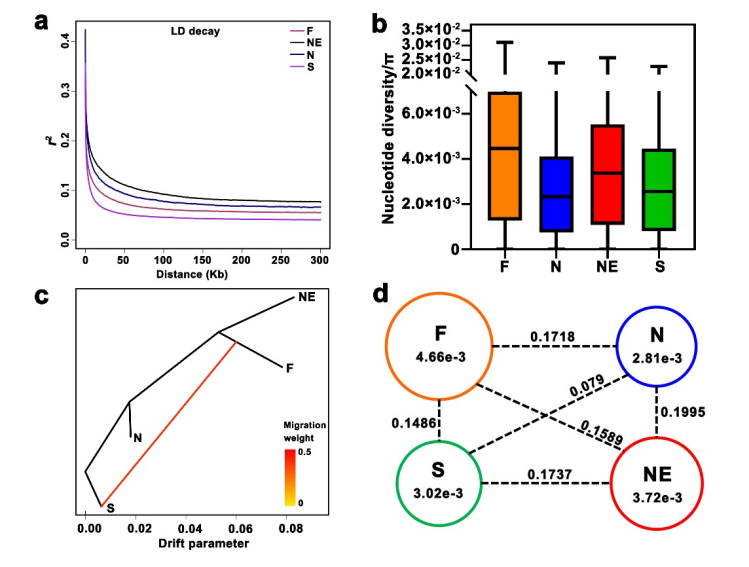
Fig. 2 The population genomic characteristics of Chinese plums.
Chinese plums exhibit strong adaptability, as they are can be cultivated both in the cold regions (e.g. Heilongjiang Province) and the subtropical regions (e.g. Guangdong Province). We exploited a combined strategy integrating nucleotide diversity (π-ratio), F ST, and XP-CLR to identify reliable regions containing selective sweeps during domestication or improvement. Pairwise comparisons between the different sub-groups identified 35 putative selection sweeps, encompassing 4.6 Mb of the P. salicina genome and involving 346 genes, among which important genes related to flowering, stress tolerance, and secondary metabolism were significantly enriched, suggesting the possible molecular basis of the domestication of these traits.
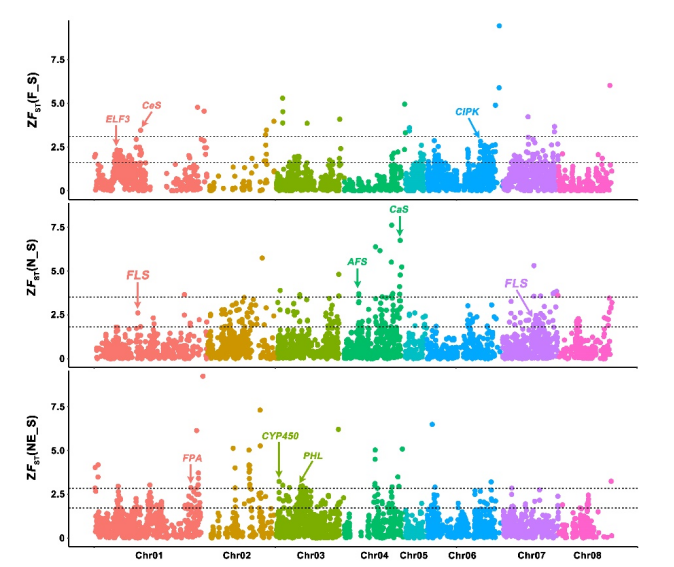
Fig. 3 Selective sweep analysis between different cultivar groups.
This work was supported by the National Key Research and Development Program (2019YFD1000203), the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-ZFRI), the National Horticulture Germplasm Resources Center, and the Key Scientific and Technological Project of Henan Province (No.202102110049).
More details are available at the following link: